Introduction
Cancers consist of a continuously evolving heterogeneous cell mass (McGranahan & Swanton, 2017). Importantly, not all cells within a tumour contribute equally to its progression. Early studies on mouse mammary tumours revealed that cellular subpopulations from different regions of the same tumour vary in growth rate, drug response, immunogenicity and metastatic capacity (reviewed in Heppner, 1984; Tabassum & Polyak, 2015). This intra‐tumour heterogeneity can arise from both genetic and non‐genetic variability within tumours, such as variations in availability of resources, like differential access to oxygen and nutrients (Kreso & Dick, 2014; Tabassum & Polyak, 2015). The development of preclinical model systems phenocopying tumour heterogeneity is required for studying its contribution to tumour progression and acquisition of therapy resistance. Whereas the first patient‐derived tumour xenograft (PDTX) models were successfully established during the fifties (Toolan, 1953), patient‐derived tumour organoid (PDTO) models have been established only during the last decade (Sato et al, 2011) (Fig 1). Both PDTX and PDTO model systems are able to recapitulate the intra‐ and inter‐tumour heterogeneity seen in human cancers (Beckhove et al, 2003; Guenot et al, 2006; Huang et al, 2015; van de Wetering et al, 2015; Bruna et al, 2016; George et al, 2017; Nanki et al, 2018; Sachs et al, 2018; Yan et al, 2018). Therefore, these models are promising tools to study sub‐clonal dynamics within individual tumours during progression and therapy resistance (Shi et al, 2014).

Figure 1.Timeline PDTX and PDTO development
Due to the complexity of human tumours, response to clinical cancer treatments varies substantially. Additionally, mechanisms of tumour progression are poorly defined as well as drug efficacy and resistance. While a high number of anti‐cancer compounds tested for clinical safety in Phase I studies progress to Phase II efficacy testing, most of these compounds fail in Phase II and III studies, which examine the power of pharmacological responses (Dimasi et al, 2013). Such high failure rates in clinical trials headline the need of preclinical efficacy models for improved predictions of clinical outcome. Several human preclinical models are currently used, including cancer cell lines, PDTX and PDTO cultures (Figs 1 and 2). These models have improved our understanding of the mechanisms of cancer progression and provided valuable tools for the development of novel cancer treatments. Additionally, these preclinical models are used to predict clinical response to anti‐cancer agents. In this review, we discuss PDTX and PDTO model systems and compare their promises and challenges in cancer research.

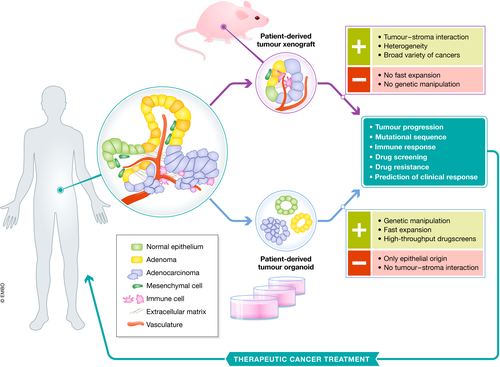
Figure 2.Schematic representation of patient‐derived tumour xenografts and organoids
PDTX models
To understand cancer biology and its translation into effective treatments, human preclinical models capturing the heterogeneity of cancer are fundamental. Although with low efficiency, primary tumour tissues can be grown in 2D cultures in vitro, allowing tumour cells capable of adapting to these conditions to expand and form a cell line. The use of in vitro cancer cell lines has provided valuable insights on tumour development and mechanisms of therapeutic actions (Sos et al, 2009; Greshock et al, 2010). However, the main drawback of cancer cell lines is the lack of both phenotypic and genetic heterogeneity found in the original tumours (Sachs & Clevers, 2014; Byrne et al, 2017). To enhance the correlation with human cancers, surgically derived primary clinical tumour samples can be grafted into mice, known as PDTX. In such models, tumour architecture and the relative proportion of cancer cells and stromal cells are maintained to a large extent, which yields better resemblance to the original tumour compared to cancer cell lines (Byrne et al, 2017; Fig 2). Although not all patient‐derived tumours can successfully be engrafted into mice, the success rate of PDTX establishment is increasing due to the establishment of immunocompromised mice (Shultz et al, 2005; Drake et al, 2012).
Although the first documented attempt to transplant a human cancer into an animal dates back to 1775, a hallmark study by Helene Toolan showed that it was possible to grow human tumour cells in x‐irradiated mice and rats (Toolan, 1953). Additionally, she demonstrated that proliferation extended considerably when the x‐radiated hosts were treated with the immune system suppressor cortisone (Toolan, 1953). Later, Phillips and Gazet were able to obtain a slightly higher percentage of viable patient‐derived tumour grafts by treating the host mice with anti‐lymphocyte serum. The number of viable grafts increased in particular when combined with thymectomy, demonstrating that a suppressed immune response enhances engraftment efficiency (Phillips & Gazet, 1970). Following these studies, several genetically modified mouse models have been established, which are severely immune deficient, such as the NOD/SCID/IL2Rγnull (NSG) mouse (Shultz et al, 2005). The virtual absence of an immune system in these mice allows for significantly higher engraftment rates (Shultz et al, 2005; Byrne et al, 2017). Together these studies demonstrated that loss of immune system activity improves engraftment and viability of patient‐derived tumour tissue into mice. PDTX models are currently established for a broad variety of cancers, including colorectal (Fichtner et al, 2004; Guenot et al, 2006), pancreatic (Fu et al, 1992; Kim et al, 2009), breast (Beckhove et al, 2003; Bruna et al, 2016), lung (Cutz et al, 2006), skin (Taetle et al, 1987), head and neck (Hennessey et al, 2011), prostate (Wang et al, 2005) and ovarian cancer (George et al, 2017; Fig 3). Although PDTXs recapitulate tumour tissue more closely than cancer cell lines, they are usually generated from a small amount of tumour material. As a consequence, the PDTX derived from it might not capture the full heterogeneity of the original tumour (Kemper et al, 2015). Moreover, Morgan and colleagues recently reported that from the total number of mutations detected in primary non‐small‐cell‐lung cancer (NSCLC) tumours, only 43% were detected in the corresponding PDTXs and four additional mutations arose in early passages of PDTXs that were not present in the primary tumour (Morgan et al, 2017). These observations suggest that clonal selection and evolution may occur early on upon tumour tissue engraftment into mice. Multiple biopsies from different regions of a tumour should be engrafted to capture the complete tumour architecture in vivo, whereas early passages of PDTXs should be used for translational applications to avoid outcomes that deviate from clinical response. Furthermore, the limited engraftment rates of PDTXs remain a major challenge, which is highly variable among cancers (Rosfjord et al, 2014). Sub‐clones of advanced tumours grow best as PDTX, compared to less advanced tumours. Additionally, growth rates of engrafted tumour tissue increase over several passages of PDTXs and a significant correlation was found between PDTX passage number and features of higher tumour grade (Pearson et al, 2016). This indicates that clonal selection occurs over passages of PDTXs.

Figure 3.Pie chart with the different cancer types that can be grown as PDTX (left) and PDTO (right) marked in green
The contribution of tumour stroma to tumour growth upon engraftment remains controversial in PDTX models. Components of human stroma, including vasculature, immune cells and fibroblasts, are present during early passages of PDTXs. The presence of these human stromal components allows for interaction studies between tumour cells and their microenvironment. However, the human stroma is subsequently replaced by murine stroma over several passages of PDTXs (Julien et al, 2012; Peng et al, 2013). Gene expression studies of NSCLC PDTXs confirmed depletion of human‐derived tumour‐associated cells with a downregulation of genes corresponding to cell adhesion and immune response pathways. This suggests that the PDTX deviates from the original tumour over time (Morgan et al, 2017). In addition, drug metabolism and pharmacokinetics differ between mouse and human, which needs to be taken into account (Morgan et al, 2017).
Over the years, it has become increasingly clear that orthotopic transplantation provide a more physiological PDTX than heterotopic (e.g. subcutaneous) engraftment. It was previously demonstrated that orthotopic transplantation can lead to local invasive growth and metastases, similar to those observed in patients (Dai et al, 2015; Hoffman, 2015). In orthotopic PDTXs, tumour‐host interactions can be investigated at the relevant location of primary and secondary tumour growth, as well as the development of metastases. In a comparison between orthotopic and subcutaneous xenografts of pancreatic ductal adenocarcinoma (PDAC), metabolic differences were found. These differences could be attributed to differences in tumour microenvironment caused by the different location of engraftment (Zhan et al, 2017). These results highlight the complexity of cancers as well as the importance of location and environment of the transplantation site. Nonetheless, while orthotopic PDTXs more accurately mimic the primary tumours by resemblance of the native microenvironment, this method is technically challenging and labour‐intensive. Therefore, most studies still use subcutaneous engraftment of tumour tissue.
PDTO models
During the last decade, techniques to grow tissues in vitro in 3D as organotypic structures have been established. These so‐called organoids can be grown from adult and embryonic stem cells and are able to self‐organize into 3D structures that reflect the tissue of origin (for adult stem cell‐derived organoids), or to which the differentiation was directed (embryonic stem cell‐derived organoids) (for a review see Clevers, 2016). The first adult stem cell‐derived organoid cultures were established from Lgr5‐expressing mouse intestinal stem cells that were placed in conditions mimicking the intestinal stem cell niche (Sato et al, 2009). By providing R‐spondin‐1, epidermal growth factor (EGF) and Noggin, and embedment of the cells in an extracellular matrix‐providing basement membranes extract, the Lgr5‐expressing stem cells received the signalling cues necessary to self‐renew, proliferate and form differentiated offspring, resembling the intestinal epithelium (Sato et al, 2009).
Since then, organoid cultures have been established for a variety of human tissues, including lung (Hild & Jaffe, 2016; Tan et al, 2017; Sachs et al, 2019), colon (Sato et al, 2011), stomach (Bartfeld et al, 2015), liver (Huch et al, 2015), pancreas (Boj et al, 2015), prostate (Chua et al, 2014; Karthaus et al, 2014), kidney (Jun et al, 2018; Schutgens et al, 2019) and fallopian tube (Kessler et al, 2015). Moreover, organoid culture protocols have been established for patient‐derived tumour tissue as well. Human tumour organoids have been generated from colon (Sato et al, 2011; van de Wetering et al, 2015), pancreas (Boj et al, 2015; Huang et al, 2015), prostate (Gao et al, 2014; Drost et al, 2016), breast (Sachs et al, 2018), gastric (Nanki et al, 2018; Yan et al, 2018), lung (Sachs et al, 2019), oesophageal (Li et al, 2018), bladder (Lee et al, 2018; Mullenders et al, 2019), ovarian (Kopper et al, 2019), kidney (Schutgens et al, 2019) and liver (Broutier et al, 2017; Li et al, 2019) tumour tissue (Fig 3). An important feature of a number of these PDTOs is that they genetically and phenotypically mirror the tumour epithelium, including its intra‐tumour heterogeneity (Huang et al, 2015; van de Wetering et al, 2015; Nanki et al, 2018; Sachs et al, 2018; Yan et al, 2018). In a recent study, Roerink and colleagues characterized organoids derived from single cells from several colorectal cancers (CRC) and showed extensive mutational diversification as well as differences in responses to anti‐cancer drugs between even closely related cells of the same tumour (Roerink et al, 2018). PDTO models show improved resemblance to the original tumour compared to 2D cultured cancer cell lines. Thereby, organoid cultures bridge the gap between in vitro 2D cancer cell line cultures and in vivo PDTXs (Sachs & Clevers, 2014; Drost & Clevers, 2018). Importantly, they can be expanded long term and cryopreserved, allowing for the generation of living tumour organoid biobanks (Weeber et al, 2015; van de Wetering et al, 2015; Fujii et al, 2016; Schütte et al, 2017; Li et al, 2018; Nanki et al, 2018; Sachs et al, 2018, 2019; Seino et al, 2018; Tiriac et al, 2018; Yan et al, 2018). So far, the majority of established PDTO cultures originate from epithelial cancers (carcinomas). Although most common adult cancers are carcinomas and epithelial in origin, a number of cancers are not, such as sarcomas, leukaemia and lymphomas. This remains a major challenge in organoid technology and is in contrast to PDTX models, which allow for the growth of a broad variety of cancers. While organoid cultures cannot mimic vasculature and tumour–stroma interactions, patient‐derived tumour organoids are a promising tool for several translational applications, such as high‐throughput drug screens and personalized medicine in a patient‐derived manner.
Translational applications of PDTX and PDTO model systems
Most preclinical anti‐cancer agents entering clinical trials fail to acquire regulatory approval due to insufficient safety or inefficacy. This highlights the limitations of the predictive value of current preclinical models. However, in a study where they evaluated the therapeutic relevance of PDTXs, a panel of six human small‐cell‐lung carcinoma (SCLC) xenografts was treated with topotecan, a topoisomerase I inhibitor, combinations of topotecan and the topoisomerase II inhibitor etoposide or alkylating agents ifosfamide or cisplatin at maximum tolerated dose (Némati et al, 2010). Three out of the six PDTX models showed over 90% growth inhibition when treated with topotecan alone, similar to the therapeutic response observed in Phase II clinical trials (Ardizzoni et al, 1997). Growth inhibition in the PDTX models was improved when topotecan was combined with etoposide or ifosfamide. These findings demonstrate that the established xenografts are useful for preclinical assessment of new drugs and combinations of drugs (Némati et al, 2010; Rosfjord et al, 2014). Moreover, Bertotti and colleagues screened a cohort of 85 metastatic CRC (mCRC) PDTX models, treated with cetuximab, an inhibiting antibody against epidermal growth factor receptor (EGFR). They found an enrichment of tumours with HER2 amplification in cetuximab‐resistant KRAS/NRAS/BRAF/PIK3CA wild‐type tumours (Bertotti et al, 2011). This proof‐of‐concept study revealed that the combined inhibition of EGFR and HER2 induced long‐lasting tumour regression, suggesting promising therapeutic opportunities for mCRC patients that are resistant to cetuximab (Bertotti et al, 2011).
Gao and colleagues generated an extensive collection of more than 1,000 PDTX models representing a broad range of solid cancers. In this large panel of PDTX models, genetic hypotheses and biomarkers of sensitivity to cancer treatments, derived from cultured cancer cell lines, were successfully validated. Importantly, the PDTX models also identified therapeutic candidates that cancer cell lines failed to capture (Gao et al, 2015; Byrne et al, 2017). The promising results of such large cohort studies increase the use of PDTXs for preclinical models of testing anti‐cancer drugs and to unravel biomarkers for drug sensitivity and resistance. For example, a recent study demonstrated that metformin, an anti‐diabetic drug, also affects tumour growth in CRC PDTX models. Administering metformin at physiological levels of 150 mg/kg per day in mice, which is equivalent to the clinical dose of 500–1,000 mg/daily in human, is sufficient to inhibit tumour growth in CRC PDTX. This implies promising therapeutic options for CRC patients (Suhaimi et al, 2017). Together, these studies demonstrate the promises of PDTXs as preclinical models to develop novel cancer treatments and predict their clinical response in patients.
Fast expansion of preclinical model systems is important to enable high‐throughput drug screens. In contrast to PDTXs, patient‐derived organoid cultures can be more easily expanded long term and several studies demonstrated that organoid cultures allow for the detection of gene–drug associations and enable high‐throughput drug screens. Verissimo and colleagues tested KRAS pathway inhibitors and combinations of drugs on normal colon organoids and CRC PDTOs and demonstrated that only organoids harbouring KRAS mutations were resistant to the treatments (Verissimo et al, 2016). In another recent study, Vlachogiannis and colleagues reported a living biobank of PDTOs from metastatic, heavily pretreated colorectal and gastroesophageal tumours, which showed a high degree of similarity to the original patient tumours. A comparison of responses to anti‐cancer agents in PDTOs and PDTO‐based orthotopic mouse tumour xenograft models with the responses of the patients in clinical trials, suggests that PDTOs successfully recapitulate the response seen in the patient (Vlachogiannis et al, 2018). Additionally, Tiriac and colleagues generated a pancreatic cancer PDTO library that largely maintained the mutational spectrum and transcriptional subtypes of primary pancreatic cancer. They showed that pancreatic cancer PDTOs exhibited heterogeneous responses to standard‐of‐care chemotherapeutics and that these therapeutic profiles correspond to patient outcomes. These data suggest that combined molecular and therapeutic profiling of PDTOs may predict treatment response and enable prospective therapeutic selection (Tiriac et al, 2018). Moreover, Schütte and colleagues collected a large biobank of 106 CRCs, 35 PDTOs and 59 PDTXs to identify novel biomarkers by linking molecular profiles with drug sensitivity patterns. Although the genetic landscape of the original tumours was largely maintained, they also found some differences between PDTXs and PDTOs, as PDTXs appeared closer to the molecular distinct CRC groups than PDTOs. Additionally, PDTOs showed elevated expression levels of genes involved in xenobiotic and fatty acid processes, which may affect drug sensitivity (Schütte et al, 2017).
Organoid cultures additionally allow for genetic engineering to study effects of oncogenic mutations in detail. Li and colleagues introduced oncogenic mutations into primary organoids from mouse colon, stomach and pancreas. This study shows that pancreatic and gastric organoids presented dysplasia as a result of the activating KrasG12D mutation, loss of Tp53 or both and formed adenocarcinoma upon in vivo transplantation. In contrast, primary colon organoids required combinatorial Apc, Tp53, KrasG12D and Smad4 mutations for the formation of adenocarcinoma in vivo. Opposed to colon organoids, small intestine organoids showed more rapid dysplasia even with only the combination of mutated Apc and Kras or mutated Apc and Tp53 (Li et al, 2014). Subsequently, two studies translated this to the human situation by CRISPR/Cas9‐mediated genome editing of common CRC driver mutations in healthy human small intestinal and colonic organoids (Drost et al, 2015; Matano et al, 2015). These studies demonstrated that organoids harbouring an activating mutation in KRAS, in combination with inactivating mutations in APC, TP53 and SMAD4, are able to grow independent of the intestinal stem cell niche factors EGF, Wnt, R‐spondin and Noggin. Additionally, Drost et al showed that loss of APC and TP53 are key drivers of chromosome instability and aneuploidy (Drost et al, 2015). Not only adult stem cell‐derived organoids can be used to study cancer initiation and progression, but also embryonic stem cell‐derived organoids can be valuable tools in cancer research. Huang et al directed the differentiation of human embryonic stem cells towards pancreatic progenitor cells that formed ductal and acinar structures. Expression of mutant KRAS or TP53 in progenitor organoids induced mutation‐specific phenotypes. For instance, mutated TP53, but not mutated KRAS, induced cytosolic SOX9 localization, which was associated with mortality of patients (Huang et al, 2015). In another recent study, Drost et al used CRISPR‐modified human stem cell organoids to study DNA repair defects in cancer (Drost et al, 2017). This showed that accumulation of mutations in organoids deficient in the mismatch repair gene MLH1 accurately models the mutation profiles observed in mismatch repair‐deficient CRC. Application of this approach to the cancer predisposition gene NTHL1 demonstrated that a high contribution of a mutational footprint (signature 30), observed in a breast cancer cohort, within a tumour can be indicative of germline mutations in NTHL1 (Drost et al, 2017). These studies demonstrate the immense opportunities organoid technology gives to study the effects of specific genetic alterations during cancer initiation and progression.
Integrating the tumour environment in PDTX and PDTO
The location in which a tumour resides, the tumour niche or microenvironment, plays an important role in cancer development. Stromal cells not only modulate the behaviour of tumour cells directly but are also able to influence the immune system (Tauriello et al, 2018). This is effectively shown in a study by Batlle and colleagues, using a mouse model in which the main four CRC driver mutations can be specifically modified in intestinal stem cells. Quadruple‐mutant mice developed metastatic tumours in the small and large intestine that showed hallmarks of human CRC, including T‐cell exclusion and TGFβ‐activated stroma. They showed that inhibition of TGFβ induced a cytotoxic T‐cell response against tumour cells that prevented metastasis, highlighting the importance of the tumour microenvironment (Tauriello et al, 2018).
Immune cells recognize antigens present on cell membranes and distinguish between cancer cells and non‐cancer cells (Schumacher & Schreiber, 2015). As a consequence of tumour‐specific mutations, cancer cells start expressing neoantigens on their membranes, which can be recognized by T lymphocytes. The recognition of such neoantigens is an important factor in the efficacy of clinical immunotherapies. Additionally, the neoantigen load may form a biomarker for cancer cells (Schumacher & Schreiber, 2015). However, the lack of an immune competent environment in both immune‐deficient mice and organoid cultures limits the utility of these models to explore the interaction between a tumour cell and the immune system. To overcome this limitation in PDTX models, humanized mouse models have been developed (Box 1). For this, selected immune components were introduced to establish a competent human immune system (HIS) in mice. Humanized mice maintain various lineages of human blood cells throughout the lifetime of the recipient animal. Ideally, the hematopoietic stem cells (HSCs) come from the same patient from whom the PDTX will be established, in order to avoid immune reactions caused by human leucocyte antigen mismatch. This is challenging, because bone marrow biopsies are a burden for weakened patients. Additionally, growth factor‐stimulated bone marrow mobilization for collecting HSCs from peripheral blood might support tumour progression (Voloshin et al, 2011). The low yields of CD34‐positive HSCs obtainable from cancer patients strongly limit the number of humanized mice. Nonetheless, humanized mice allow studying features of the human anti‐tumour immune response in a mouse model system (Shultz et al, 2012; Byrne et al, 2017). In such a study, newborn NSG mice were co‐engrafted with human HSCs and human breast cancer cells. In these mice, tumour growth was accompanied by specific T‐cell maturation as well as tumour cell‐specific activation of T cells. Additionally, an accumulation of NK cells was observed at the tumour site (Wege et al, 2011). Transplantation of primary lung tumours into humanized mice revealed the existence of tumour‐infiltrating effector memory T cells that were activated upon human IL‐12 administration (Simpson‐Abelson et al, 2008). The ability to study tumour progression combined with its engrafted immune system provides new approaches for cancer immunotherapy, resistance of tumour cells to anti‐cancer therapies and the involvement of the immune system in response to chemotherapy.
In tumour organoid cultures, the lack of an immune competent environment can be overcome by co‐culture with immune cells. Nozaki and colleagues developed a novel co‐culture system of mouse intra‐epithelial lymphocytes and intestinal epithelial cells. In this co‐culture, the intra‐epithelial lymphocytes were expanded with intestinal organoids in the presence of IL‐2, IL‐7 and IL‐15 (Nozaki et al, 2016). Recently, Zumwalde et al (2016) succeeded in characterizing the intra‐epithelial lymphocyte compartment of healthy human breast tissue as well as identifying a subset of T lymphocytes that can be pharmacologically targeted to enhance their response to breast cancer cells. Specifically, Vδ2+ γδ T cells were constantly present in the preparation of mammary ductal epithelial organoids. In response to zoledronic acid, an aminobisphosphonate drug, these T lymphocytes started to proliferate. Additionally, Vδ2+ T cells from breast ductal organoids produced IFNγ, an anti‐tumour cytokine, and efficiently killed bisphosphonate‐pulsed breast cancer cells. Together, these results demonstrate the potential for Vδ2+ γδ T lymphocytes to respond to FDA‐approved bisphosphonate drugs as a novel immunotherapeutic approach to inhibit cancer growth (Zumwalde et al, 2016). In another recent study, Dijkstra and colleagues established and validated a platform to induce and analyse tumour‐specific T‐cell responses to epithelial cancers, including mismatch repair‐deficient CRC and NSCLC, in a personalized manner. Enrichment of tumour‐reactive T cells from peripheral blood of patients was successfully established by co‐cultures of peripheral blood lymphocytes with autologous tumour organoids. Moreover, they demonstrated that these tumour‐reactive T cells efficiently recognize and kill autologous tumour organoids, while leaving the healthy organoids or tissue unharmed (Dijkstra et al, 2018). In addition, Neal et al (2018) developed an air–liquid interface PDTO culture system that recapitulates complex tumour architecture including stromal and immune compartments. They demonstrated that the T‐cell receptors are highly conserved between the PDTO culture and the parental tumour. Crucially, they showed that the PDTO cultures functionally recapitulate the PD‐1/PD‐L1‐dependent immune checkpoint (Neal et al, 2018).
In addition to the immune system, cancer‐associated fibroblasts (CAFs) play an important role in the tumour environment. As such, Öhlund and colleagues showed that a co‐culture of murine pancreatic stellate cells (PSCs) and PDAC tumour organoids recapitulate properties of PDAC desmoplasia. They demonstrate that PSCs differentiate into two distinct subtypes of CAFs with elevated expression of αSMA and secretion of IL‐6 and additional inflammatory mediators, respectively (Öhlund et al, 2017). In accordance with this, Seino and colleagues established a co‐culture of PDAC organoids and CAFs and showed that the CAFs provide a WNT niche for PDAC (Seino et al, 2018), highlighting the importance of CAFs in the tumour microenvironment. In an effort to model diabetic vasculopathy in vitro, a recent study reported the development of human blood vessel organoids from pluripotent stem cells that self‐assemble into capillary networks, containing endothelial cells and pericytes, surrounded by a basement membrane. Upon transplantation of these organoids into mice, a perfused vascular tree is formed, including arteries, arterioles and venules (Wimmer et al, 2019). These human blood vessel organoids may open new doors for PDTO co‐cultures. Culturing PDTOs in the presence of the vascular system, in addition to co‐cultures with CAFs and immune cells, can recapitulate more components of the tumour microenvironment in vitro.
In conclusion, the lack of an immune competent environment in both PDTX and PDTO model systems can be overcome by using humanized mouse models or generating co‐cultures with immune cells, respectively. Additionally, to mimic the tumour stroma in the organoid model system, PDTOs can be co‐cultured with CAFs.
Box 1: Humanized mouse models
Humanized mice are immunodeficient mice engrafted with human hematopoietic stem cells which give rise to a variety of human blood cell lineages throughout the life of the animal. The human immune system (HIS) mouse model can be generated by transplantation of CD34‐positive human hematopoietic stem cells (HSCs) or precursor cells. These cells can be isolated from bone marrow, peripheral blood or umbilical cord blood. CD34‐positive HSC transplantation can be performed alone or in combination with transplantation of human immune tissues, such as thymic tissue (Drake et al, 2012). Humanized mouse models are powerful tools for studying cancer, haematopoiesis, and inflammatory and infectious disease.
Drug screens and personalized medicine using PDTX and PDTO models
The discovery of molecular biomarkers for drug sensitivity is of high importance for the treatment of cancer patients. As illustrated by the EGFR tyrosine kinase inhibitor gefitinib in NSCLC, some drugs can give exceptional responses in small subsets of patients. However, when these patients are not properly identified within larger cohorts, it results in an overall negative clinical trial outcome (Thatcher et al, 2005). PDTX models can be used as screening platforms for predicting clinical outcome of the response rate to drugs, identifying biomarkers for drug sensitivity and studying drug resistance. For example, a prospective study in PDAC showed that the combination of the anti‐microtubule agent nab‐paclitaxel and the anti‐metabolite gemcitabine is effective in PDTX models of PDAC, which correlated with the clinical efficacy of this combination. Moreover, this combination of chemotherapeutics has been demonstrated to provide a survival benefit for advanced PDAC patients in a randomized phase III study (Von Hoff et al, 2013; Hidalgo et al, 2015). Additionally, PDTX models are not only able to provide potential clinical indications, but they may also facilitate the identification of potential drug efficacy biomarkers. In CRC for example, several studies have shown that KRAS mutant PDTX models do not respond to cetuximab. The wild‐type status of KRAS is now a well‐documented clinical biomarker for this targeted therapy (Hidalgo et al, 2015). Furthermore, melanoma PDTX models were involved in the identification of a mechanism of resistance to targeted drugs, such as the BRAFV600E inhibitor vemurafenib. Additionally, a novel drug administration strategy that is clinically applicable was proposed to overcome resistance (Das Thakur et al, 2013). Although PDTX models are useful for low‐throughput drug screens and predicting clinical outcome, they do not allow for high‐throughput drug screens.
In contrast to PDTX models, PDTOs can be established and expanded more efficiently, which allows them to be used in medium‐ to high‐throughput drug screens. The use of PDTOs as a preclinical model for finding biomarkers and performing genotype–drug associations is just starting to be explored. A few studies already show promising results. For example, drug testing on a panel of organoids derived from 20 chemo‐naive CRC patients confirmed known drug sensitivity–genotype correlations. This proof of principle highlighted the potential of screening organoid biobanks to detect novel genotype–phenotype correlations (van de Wetering et al, 2015). In a recent study, Pauli and colleagues collected 145 specimens, representing 18 different tumour types derived from patients with metastatic solid tumours. PDTO cultures were established from 56 of these specimens (38.6%), which were obtained from biopsies or surgical resection and stored in a living biobank (Pauli et al, 2017). PDTOs of four patients were used to perform drug screens, targeting mutated pathways that were identified using whole exome sequencing (WES). PDTX models were subsequently used for the validation of the compounds that affected PDTO growth. They found that two patients, suffering from uterine carcinosarcoma and endometrial adenocarcinoma, respectively, carried similar driver mutations in PIK3CA and PTEN. Yet, the drug response profiles clearly distinguished the two patients. For the uterine carcinosarcoma case, they identified the combination of the PIK3 inhibitor buparlisib (Armstrong et al, 2017) with the hypoxia signalling suppressor vorinostat (Zhang et al, 2017) as one of the top drug combinations. By contrast, for the endometrial adenocarcinoma case, a combination of buparlisib with the PARP and HDAC inhibitor olaparib (Yuan et al, 2017) was found as optimal treatment in both PDTO and PDTX models. The latter is of high relevance, as no targeted therapies are approved for endometrial cancer yet (Pauli et al, 2017). The combination of WES, PDTO and PDTX models makes it possible to compare the efficacy of specific drugs on individual tumours thereby providing recommendations for patient care in a personalized manner. Moreover, it enables assessing how individual tumours evolve in response to therapies as well as determining next therapeutic steps for cases where standard clinical options have already been exhausted. Additionally, the combination of these techniques allows creating a database that relates drug sensitivity to tumour genetics. This enables to nominate potential therapeutic strategies even when only genomic data are available (Pauli et al, 2017). In conclusion, combining different techniques and model systems for cancer research can improve the prediction of clinical response to anti‐cancer treatments in a personalized manner.
Outlook and challenges
The main challenge in preclinical cancer research remains the establishment of models that recapitulate the patient situation as close as possible, retaining intra‐tumour heterogeneity and the tumour environment. So far, not all cancer types can be grown in mice or as tumour organoids. For the establishment of more PDTO models, it is of high relevance to find the optimal in vitro growth conditions that enable tumour cells to grow, while keeping as much cellular heterogeneity as possible. Selection pressure occurs in both PDTX and PDTO model systems (Morgan et al, 2017; Pauli et al, 2017). Therefore, combining the strengths of both preclinical model systems will be powerful for investigating therapeutic responses. For example, PDTOs can be used for high‐throughput drug screenings and selecting effective drugs or drug combinations. Subsequently, the efficacy of these selected drugs should be validated in PDTX models (Pauli et al, 2017). Together, these preclinical models can reflect the response to anti‐cancer therapies and give indications for patient‐tailored treatment.
Conclusions
In this review, we discussed the role of PDTX and PDTO model systems in cancer research and therapy development (Table 1). While PDTXs have already been established for a broad variety of cancers (Fig 3), the fast‐evolving improvements in PDTO culture systems hold great promise. Transplanting primary tumour tissue directly into mice allows for the partial resemblance of the tumour mirco‐environment, including stromal components, such as CAFs and vasculature. Although PDTO cultures do—so far—not maintain the stromal components of human tumours, they represent genetic and phenotypic heterogeneity found in human cancers. Additionally, organoid cultures can be expanded relatively fast, cryopreserved and genetically modified. These features allow for generating living tumour organoid biobanks and providing a platform for high‐throughput drug screens. Although both models lack an immune competent environment, this limitation can be overcome by transplantation of HSCs and co‐culture with T lymphocytes for PDTX and PDTO models, respectively.
Characteristics of patient‐derived tumour xenograft (PDTX) and tumour organoid (PDTO) model systems. Features are rated as best (++), suitable (+), possible (−/+) and unsuitable (−) (adapted from Sachs & Clevers, 2014)
| Feature | PDTX | PDTO |
|---|---|---|
| Ease of use | + | ++ |
| Initiation efficiency | + | ++ |
| Scalability | + | ++ |
| Genetic stability | ++ | ++ |
| Intra‐tumour heterogeneity | ++ | + |
| Genetic modification | −/+ | ++ |
| Integratable immune system | −/+ | −/+ |
| Tumour–stroma interaction | + | −/+ |
| Low‐throughput drug screens | + | ++ |
| High‐throughput drug screens | − | ++ |
| Prediction of clinical response | ++ | ++ |
| Testable drug classesa | 3 | 2 |
- a
Drug classes: (1) agents targeting tumour‐specific proteins, (2) agents targeting host–tumour interactions and (3) agents targeting tumour cells empirically.
In addition to PDTX and PDTO, several other preclinical models have been developed over the years. Induced pluripotent stem cells (iPSCs) can be differentiated towards several lineages and can be used to model normal development in vitro. iPSCs were successfully used for exocrine differentiation of pancreatic progenitors and for modelling PDAC tumour organoids (Huang et al, 2015) as well as for differentiation to colonic organoids for the modelling of CRC (Crespo et al, 2018). However, two main challenges in the establishment of iPSC‐derived cancer models are the efficiency of malignant cell reprogramming and the capability to differentiate the iPSCs into the cell lineage of interest (Papapetrou, 2016). Additionally, during the establishment of iPSC‐derived cancer models, selective outgrowth of tumour sub‐clones may occur when a subset of cells harbours specific mutations resulting in a loss of heterogeneity. This is in stark contrast to PDTO model systems that largely recapitulate the genetic heterogeneity of the parental tumour (Huang et al, 2015; van de Wetering et al, 2015; Nanki et al, 2018; Sachs et al, 2018; Yan et al, 2018). Another recent study described a technology termed conditional reprogramming, which allows efficient establishment of patient tissue‐derived 2D cancer cell cultures in the presence of RHO kinase inhibitor and a fibroblast feeder layer (Liu et al, 2012). It is of high importance to combine multiple cancer models to get the best possible prediction of tumour sensitivity to and toxicity of anti‐cancer treatments, which will ultimately result in more efficient translation from bench to bedside.
Acknowledgements
This review was prepared with support from the Dutch Cancer Society (KWF)/Alpe d’HuZes Bas Mulder Award to J.D. (KWF/Alpe d’HuZes, grant #10218), and Oncode Institute to H.C. and J.D.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1. Ardizzoni BA, Hansen H, Dombernowsky P, Gamucci T, Kaplan S, Postmus P (1997) Topotecan, a new active drug in the second‐line treatment of small‐cell lung cancer : a phase ii study in patients with refractory and sensitive disease. J Clin Oncol 15: 2090– 2096
Crossref
CAS
PubMed
Web of Science®Google Scholar - 2. Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J, Bitting RL, Hobbs C, Soleau CF, Beer TM et al (2017) Phase II trial of the PI3 kinase inhibitor buparlisib (BKM‐120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer 81: 228– 236
Crossref
CAS
PubMed
Web of Science®Google Scholar - 3. Bartfeld S, Bayram T, Van De Wetering M, Huch M, Begthel H, Kujala P, Vries R, Peters PJ, Clevers H (2015) In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148: 126– 136
Crossref
PubMed
Web of Science®Google Scholar - 4. Beckhove P, Schütz F, Diel IJ, Solomayer EF, Bastert G, Foerster J, Feuerer M, Bai L, Sinn HP, Umansky V et al (2003) Efficient engraftment of human primary breast cancer transplants in nonconditioned NOD/SCID mice. Int J Cancer 105: 444– 453
Wiley Online Library
CAS
PubMed
Web of Science®Google Scholar - 5. Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, di Nicolantonio F, Buscarino M, Petti C et al (2011) A molecularly annotated platform of patient‐ derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab‐resistant colorectal cancer. Cancer Discov 1: 508– 523
Crossref
CAS
PubMed
Web of Science®Google Scholar - 6. Boj SF, Il Hwang C, Baker LA, Chio IIC, Engle DD, Corbo V, Jager M, Ponz‐Sarvise M, Tiriac H, Spector MS et al (2015) Organoid models of human and mouse ductal pancreatic cancer. Cell 160: 324– 338
Crossref
CAS
PubMed
Web of Science®Google Scholar - 7. Broutier L, Mastrogiovanni G, Verstegen MMA, Francies HE, Gavarró LM, Bradshaw CR, Allen GE, Arnes‐benito R, Sidorova O, Gaspersz MP et al (2017) Human primary liver cancer – derived organoid cultures for disease modeling and drug screening. Nat Med 23: 1424– 1435
Crossref
CAS
PubMed
Web of Science®Google Scholar - 8. Bruna A, Rueda OM, Caldas C (2016) Modeling breast cancer intertumor and intratumor heterogeneity using xenografts. Cold Spring Harb Symp Quant Biol 81: 227– 230
CrossrefGoogle Scholar - 9. Byrne AT, Alférez DG, Amant F, Annibali D, Arribas J, Biankin AV, Bruna A, Budinská E, Caldas C, Chang DK et al (2017) Interrogating open issues in cancer precision medicine with patient‐derived xenografts. Nat Rev Cancer 17: 254– 268
Crossref
CAS
PubMed
Web of Science®Google Scholar - 10. Chua CW, Shibata M, Lei M, Toivanen R, Barlow LJ, Bergren SK, Badani KK, Mckiernan JM, Benson MC, Hibshoosh H et al (2014) Single luminal epithelial progenitors can generate prostate organoids in culture. Nat Cell Biol 16: 951– 961
Crossref
CAS
PubMed
Web of Science®Google Scholar - 11. Clevers H (2016) Modeling Development and Disease with Organoids. Cell 165: 1586– 1597
Crossref
CAS
PubMed
Web of Science®Google Scholar - 12. Crespo M, Vilar E, Tsai S, Chang K, Amin S, Srinivasan T, Zhang T, Pipalia NH, Chen HJ, Witherspoon M et al (2018) Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat Med 23: 878– 884
Crossref
Web of Science®Google Scholar - 13. Cutz J, Guan J, Bayani J, Xue H, Sutcliffe M, English J, Flint J, Leriche J, Yee J, Squire J et al (2006) Cancer Therapy : Preclinical Establishment in Severe Combined Immunodeficiency Mice of Subrenal Capsule Xenografts and Transplantable Tumor Lines from a Variety of Primary. Human Lung Cancers 12: 4043– 4055
CASGoogle Scholar - 14. Dai L, Lu C, Yu X, Dai L‐J, Zhou JX (2015) Construction of orthotopic xenograft mouse models for human pancreatic cancer. Exp Ther Med 10: 1033– 1038
Crossref
CAS
Web of Science®Google Scholar - 15. Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD (2013) Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494: 251– 255
Crossref
CAS
PubMed
Web of Science®Google Scholar - 16. Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S et al (2018) Generation of tumor‐reactive T cells by co‐culture of peripheral blood lymphocytes and tumor organoids. Cell 174: 1586– 1598.e12
Crossref
CAS
PubMed
Web of Science®Google Scholar - 17. Dimasi JA, Reichert JM, Feldman L, Malins A (2013) Clinical approval success rates for investigational cancer drugs. Clin Pharmacol Ther 94: 329– 335
Wiley Online Library
CAS
PubMed
Web of Science®Google Scholar - 18. Drake AC, Chen Q, Chen J (2012) Engineering humanized mice for improved hematopoietic reconstitution. Cell Mol Immunol 9: 215– 224
Crossref
CAS
PubMed
Web of Science®Google Scholar - 19. Drost J, Van Jaarsveld RH, Ponsioen B, Zimberlin C, Van Boxtel R, Buijs A, Overmeer M, Offerhaus GJ, Begthel H, Korving J et al (2015) Sequential cancer mutations in cultured human intestinal stem cells. Nature 521: 43– 47
Crossref
CAS
PubMed
Web of Science®Google Scholar - 20. Drost J, Karthaus WR, Gao D, Driehuis E, Sawyers CL, Chen Y, Clevers H (2016) Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc 11: 347– 358
Crossref
CAS
PubMed
Web of Science®Google Scholar - 21. Drost J, Van Boxtel R, Blokzijl F, Mizutani T, Sasaki N, Sasselli V, de Ligt J, Behjati S, Grolleman JE, van Wezel T et al (2017) Use of CRISPR‐modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 91: 399– 404Google Scholar
- 22. Drost J, Clevers H (2018) Organoids in cancer research. Nat Rev Cancer 18: 407– 418
Crossref
CAS
PubMed
Web of Science®Google Scholar - 23. Fichtner I, Slisow W, Gill J, Becker M, Elbe B, Hillebrand T, Bibby M (2004) Anticancer drug response and expression of molecular markers in early‐passage xenotransplanted colon carcinomas. Eur J Cancer 40: 298– 307
Crossref
CAS
PubMed
Web of Science®Google Scholar - 24. Fu X, Guadagni F, Hoffman RM (1992) A metastatic nude‐mouse model of human pancreatic cancer constructed orthotopically with histologically intact patient specimens. Proc Natl Acad Sci USA 89: 5645– 5649
Crossref
CAS
PubMed
Web of Science®Google Scholar - 25. Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, Ohta Y, Toshimitsu K, Nakazato Y, Kawasaki K et al (2016) A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 18: 827– 838
Crossref
CAS
PubMed
Web of Science®Google Scholar - 26. Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK et al (2014) Organoid cultures derived from patients with advanced prostate cancer. Cell 159: 176– 187
Crossref
CAS
PubMed
Web of Science®Google Scholar - 27. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y et al (2015) High‐throughput screening using patient‐derived tumor xenografts to predict clinical trial drug response. Nat Med 21: 1318– 1325
Crossref
CAS
PubMed
Web of Science®Google Scholar - 28. George E, Kim H, Krepler C, Wenz B, Makvandi M, Tanyi JL, Brown E, Zhang R, Brafford P, Jean S et al (2017) A patient‐derived‐xenograft platform to study BRCA‐deficient ovarian cancers. JCI Insight 2: e89760
Crossref
Web of Science®Google Scholar - 29. Greshock J, Bachman KE, Degenhardt YY, Jing J, Wen YH, Eastman S, McNeil E, Moy C, Wegrzyn R, Auger K et al (2010) Molecular target class is predictive of in vitro response profile. Cancer Res 70: 3677– 3686
Crossref
CAS
PubMed
Web of Science®Google Scholar - 30. Guenot D, Guérin E, Aguillon‐Romain S, Pencreach E, Schneider A, Neuville A, Chenard MP, Duluc I, Du Manoir S, Brigand C et al (2006) Primary tumour genetic alterations and intra‐tumoral heterogeneity are maintained in xenografts of human colon cancers showing chromosome instability. J Pathol 208: 643– 652
Wiley Online Library
CAS
PubMed
Web of Science®Google Scholar - 31. Hennessey PT, Ochs MF, Mydlarz WW, Hsueh W, Cope L, Yu W, Califano JA (2011) Promoter methylation in head and neck squamous cell carcinoma cell lines is significantly different than methylation in primary tumors and xenografts. PLoS ONE 6: e20584
Crossref
CAS
PubMed
Web of Science®Google Scholar - 32. Heppner GH (1984) Tumor heterogeneity. Cancer Res 44: 2259– 2265
CAS
PubMed
Web of Science®Google Scholar - 33. Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, Clarke RB, De Jong S, Jonkers J, Mari G (2015) Patient derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 4: 998– 1013
Crossref
Web of Science®Google Scholar - 34. Hild M, Jaffe AB (2016) Production of 3‐D airway organoids from primary human airway basal cells and their use in high‐throughput screening. Curr Protoc Stem Cell Biol 37: IE.9.1– IE.9.15
Wiley Online LibraryGoogle Scholar - 35. Hoffman RM (2015) Patient‐derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nat Rev Cancer 15: 451– 452
Crossref
CAS
PubMed
Web of Science®Google Scholar - 36. Huang L, Holtzinger A, Jagan I, Begora M, Lohse I, Ngai N, Nostro C, Wang R, Muthuswamy LB, Crawford HC et al (2015) Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell‐ and patient‐derived tumor organoids. Nat Med 21: 1364– 1371
Crossref
CAS
PubMed
Web of Science®Google Scholar - 37. Huch M, Gehart H, Van Boxtel R, Hamer K, Blokzijl F, Verstegen MMA, Ellis E, Van Wenum M, Fuchs SA, De Ligt J et al (2015) Long‐term culture of genome‐stable bipotent stem cells from adult human liver. Cell 160: 299– 312
Crossref
CAS
PubMed
Web of Science®Google Scholar - 38. Julien S, Merino‐trigo A, Lacroix L, Pocard M, Goére D, Mariani P, Landron S, Bigot L, Nemati F, Dartigues P et al (2012) Characterization of a large panel of patient‐derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res 18: 5314– 5329
Crossref
CAS
PubMed
Web of Science®Google Scholar - 39. Jun D, Kim SY, Na JC, Ho H, Id L, Kim J, Yoon YE, Hong SJ, Kyu W, Id H (2018) Tubular organotypic culture model of human kidney. PLoS ONE 13: e0206447
Crossref
Web of Science®Google Scholar - 40. Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, Van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N et al (2014) Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 159: 163– 175
Crossref
CAS
PubMed
Web of Science®Google Scholar - 41. Kemper K, Krijgsman O, Cornelissen‐Steijger P, Shahrabi A, Weeber F, Song J‐Y, Kuilman T, Vis DJ, Wessels LF, Voest EE et al (2015) Intra‐ and inter‐tumor heterogeneity in a vemurafenib‐resistant melanoma patient and derived xenografts. EMBO Mol Med 7: 1104– 1118
Wiley Online Library
CAS
PubMed
Web of Science®Google Scholar - 42. Kessler M, Hoffmann K, Brinkmann V, Thieck O, Jackisch S, Toelle B, Berger H, Mollenkopf HJ, Mangler M, Sehouli J et al (2015) The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat Commun 6: 8989
Crossref
CAS
PubMed
Web of Science®Google Scholar - 43. Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE (2009) Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 4: 1670– 1680
Crossref
CAS
PubMed
Web of Science®Google Scholar - 44. Kopper O, De Witte CJ, Lõhmussaar K, Valle‐inclan JE, Hami N, Kester L, Balgobind AV, Korving J, Proost N, Begthel H et al (2019) An organoid platform for ovarian cancer captures intra‐ and interpatient heterogeneity. Nat Med 25: 838– 849
Crossref
CAS
Web of Science®Google Scholar - 45. Kreso A, Dick JE (2014) Review evolution of the cancer stem cell model. Stem Cell 14: 275– 291
CAS
Web of Science®Google Scholar - 46. Lee SH, Hu W, Matulay JT, Al‐ahmadie H, Solit DB, Shen MM, Lee SH, Hu W, Matulay JT, Silva MV et al (2018) Tumor evolution and drug response in patient‐ derived organoid models of bladder cancer. Cell 173: 515– 528
Crossref
CAS
PubMed
Web of Science®Google Scholar - 47. Li X, Nadauld L, Ootani A, Corney DC, Pai RK, Gevaert O, Cantrell MA, Rack PG, Neal JT, Chan CWM et al (2014) Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med 20: 769– 777
Crossref
CAS
PubMed
Web of Science®Google Scholar - 48. Li X, Francies HE, Secrier M, Perner J, Miremadi A, Galeano‐dalmau N, Barendt WJ, Letchford L, Leyden GM, Goffin EK et al (2018) Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat Commun 9: 2983
Crossref
Web of Science®Google Scholar - 49. Li L, Bader JS, Selaru FM, Li L, Knutsdottir H, Hui K, Weiss MJ, He J, Philosophe B, Ewald AJ et al (2019) Human primary liver cancer organoids reveal intratumor and interpatient drug response heterogeneity. JCI Insight 4: 121490
Crossref
Web of Science®Google Scholar - 50. Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V et al (2012) ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 180: 599– 607
Crossref
CAS
PubMed
Web of Science®Google Scholar - 51. Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, Watanabe T, Kanai T, Sato T (2015) Modeling colorectal cancer using CRISPR‐Cas9–mediated engineering of human intestinal organoids. Nat Med 21: 256– 262
Crossref
CAS
PubMed
Web of Science®Google Scholar - 52. McGranahan N, Swanton C (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168: 613– 628
Crossref
CAS
PubMed
Web of Science®Google Scholar - 53. Morgan KM, Riedlinger GM, Rosenfeld J, Ganesan S, Pine SR (2017) Patient‐derived xenograft models of non‐small cell lung cancer and their potential utility in personalized medicine. Front Oncol 7: 2
Crossref
Web of Science®Google Scholar - 54. Mullenders J, De Jongh E, Brousali A, Roosen M, Blom JPA, Begthel H, Korving J, Jonges T, Kranenburg O, Meijer R et al (2019) Mouse and human urothelial cancer organoids : a tool for bladder cancer research. Proc Natl Acad Sci USA 116: 4567– 4574
Crossref
CAS
Web of Science®Google Scholar - 55. Nanki K, Toshimitsu K, Takano A, Fujii M, Shimokawa M, Ohta Y, Matano M, Seino T, Nishikori S, Ishikawa K et al (2018) Divergent routes toward Wnt and R‐spondin niche independency during human gastric carcinogenesis. Cell 174: 856– 869.e17
Crossref
CAS
Web of Science®Google Scholar - 56. Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, Liu IH, Chiou SH, Salahudeen AA, Smith AR et al (2018) Organoid modeling of the tumor immune microenvironment. Cell 175: 1972– 1988.e16
Crossref
CAS
Web of Science®Google Scholar - 57. Némati F, Daniel C, Arvelo F, Legrier ME, Froget B, Livartowski A, Assayag F, Bourgeois Y, Poupon MF, Decaudin D (2010) Clinical relevance of human cancer xenografts as a tool for preclinical assessment: example of in‐vivo evaluation of topotecan‐based chemotherapy in a panel of human small‐cell lung cancer xenografts. Anticancer Drugs 21: 25– 32
Crossref
CAS
PubMed
Web of Science®Google Scholar - 58. Nozaki K, Mochizuki W, Matsumoto Y, Matsumoto T, Fukuda M, Mizutani T, Watanabe M, Nakamura T (2016) Co‐culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J Gastroenterol 51: 206– 213
Crossref
CAS
PubMed
Web of Science®Google Scholar - 59. Öhlund D, Santana AH, Biffi G, Elyada E, Almeida AS, Sarvise MP, Corbo V, Oni TE, Hearn SA, Lee EJ et al (2017) Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214: 579– 596
CAS
PubMed
Web of Science®Google Scholar - 60. Papapetrou EP (2016) perspective Patient‐derived induced pluripotent stem cells in cancer research and precision oncology. Nat Med 22: 1392– 1401
Crossref
CAS
PubMed
Web of Science®Google Scholar - 61. Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, Sailer V, Augello M, Puca L, Rosati R et al (2017) Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov 7: 462– 477
Crossref
PubMed
Web of Science®Google Scholar - 62. Pearson AT, Finkel KA, Warner KA, Nör F, Tice D, Martins MD, Jackson TL, Nör JE (2016) Patient‐derived xenograft (PDX) tumors increase growth rate with time. Oncotarget 7: 7993– 8005
Crossref
PubMed
Web of Science®Google Scholar - 63. Peng S, Creighton CJ, Zhang Y, Sen B, Mazumdar T, Myers JN, Lai SY, Woolfson A, Lorenzi MV, Bell D et al (2013) Tumor grafts derived from patients with head and neck squamous carcinoma authentically maintain the molecular and histologic characteristics of human cancers. J Transl Med 11: 198
Crossref
CAS
PubMed
Web of Science®Google Scholar - 64. Phillips B, Gazet JC (1970) Transplantation of primary explants of human tumour to mice treated with antilymphocyte serum. Br J Cancer 24: 92– 95
Crossref
CAS
PubMed
Web of Science®Google Scholar - 65. Roerink SF, Sasaki N, Lee‐Six H, Young MD, Alexandrov LB, Behjati S, Mitchell TJ, Grossmann S, Lightfoot H, Egan DA et al (2018) Intra‐tumour diversification in colorectal cancer at the single‐cell level. Nature 556: 437– 462
Crossref
Web of Science®Google Scholar - 66. Rosfjord E, Lucas J, Li G, Gerber HP (2014) Advances in patient‐derived tumor xenografts: from target identification to predicting clinical response rates in oncology. Biochem Pharmacol 91: 135– 143
Crossref
CAS
PubMed
Web of Science®Google Scholar - 67. Sachs N, Clevers H (2014) ScienceDirect Organoid cultures for the analysis of cancer phenotypes. Curr Opin Genet Dev 24: 68– 73
Crossref
CAS
PubMed
Web of Science®Google Scholar - 68. Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H et al (2018) Resource a living biobank of breast cancer organoids resource a living biobank of breast cancer organoids captures disease heterogeneity. Cell 172: 373– 386
Crossref
CAS
PubMed
Web of Science®Google Scholar - 69. Sachs N, Papaspyropoulos A, Ommen DDZ, Heo I, Klay D, Weeber F, Huelsz‐prince G, Iakobachvili N, Gimano D, De Ligt J et al (2019) Long‐term expanding human airway organoids for disease modeling. EMBO J 38: e100300
Wiley Online Library
Web of Science®Google Scholar - 70. Sato T, Vries RG, Snippert HJ, Van De Wetering M, Barker N, Stange DE, Van Es JH, Abo A, Kujala P, Peters PJ et al (2009) Single Lgr5 stem cells build crypt – villus structures in vitro without a mesenchymal niche. Nature 459: 262– 265
Crossref
CAS
PubMed
Web of Science®Google Scholar - 71. Sato T, Stange DE, Ferrante M, Vries RGJ, Van Es JH, Van Den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD et al (2011) Long‐term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141: 1762– 1772
Crossref
CAS
PubMed
Web of Science®Google Scholar - 72. Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Sci Rev 348: 69– 74
CASGoogle Scholar - 73. Schutgens F, Rookmaaker MB, Margaritis T, Rios A, Ammerlaan C, Jansen J, Gijzen L, Vormann M, Vonk A, Viveen M et al (2019) Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat Biotechnol 37: 303– 313
Crossref
CAS
Web of Science®Google Scholar - 74. Schütte M, Risch T, Abdavi‐Azar N, Boehnke K, Schumacher D, Keil M, Yildiriman R, Jandrasits C, Borodina T, Amstislavskiy V et al (2017) Molecular dissection of colorectal cancer in pre‐clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat Commun 8: 14262
Crossref
CAS
PubMed
Web of Science®Google Scholar - 75. Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M (2018) Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Stem Cell 22: 454– 467
CASGoogle Scholar - 76. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB et al (2014) Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 4: 80– 93
Crossref
CAS
PubMed
Web of Science®Google Scholar - 77. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J et al (2005) Human Lymphoid and Myeloid Cell Development in NOD/LtSz‐scid IL2R null Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J Immunol 174: 6477– 6489
Crossref
CAS
PubMed
Web of Science®Google Scholar - 78. Shultz LD, Brehm MA, Victor Garcia‐Martinez J, Greiner DL (2012) Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol 12: 786– 798
Crossref
CAS
PubMed
Web of Science®Google Scholar - 79. Simpson‐Abelson MR, Sonnenberg GF, Takita H, Yokota SJ, Conway TF, Kelleher RJ, Shultz LD, Barcos M, Bankert RB (2008) Long‐Term Engraftment and Expansion of Tumor‐Derived Memory T Cells Following the Implantation of Non‐Disrupted Pieces of Human Lung Tumor into NOD‐scid IL2R null Mice. J Immunol 180: 7009– 7018
Crossref
CAS
PubMed
Web of Science®Google Scholar - 80. Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, Li D, Ullrich R, Koker M, Fischer F et al (2009) Predicting drug susceptibility of non ‐ small cell lung cancers based on genetic lesions. J Clin Invest 119: 1727– 1740
Crossref
CAS
PubMed
Web of Science®Google Scholar - 81. Suhaimi N‐AM, Phyo WM, Yap HY, Choy SHY, Wei X, Choudhury Y, Tan WJ, Tan LAPY, Foo RSY, Tan SHS et al (2017) Metformin inhibits cellular proliferation and bioenergetics in colorectal cancer patient‐derived xenografts. Mol Cancer Ther 16: 2035– 2044
Crossref
Web of Science®Google Scholar - 82. Tabassum DP, Polyak K (2015) Tumorigenesis: it takes a village. Nat Rev Cancer 15: 473– 483
Crossref
CAS
PubMed
Web of Science®Google Scholar - 83. Taetle R, Jones OW, Honeysett JM, Abramson I, Bradshaw C, Reid S (1987) Use of nude mouse xenografts as preclinical screens: characterization of xenograft‐derived melanoma cell lines. Cancer 60: 1836– 1841
Wiley Online Library
CAS
PubMed
Web of Science®Google Scholar - 84. Tan Q, Choi KM, Sicard D, Tschumperlin DJ (2017) Human airway organoid engineering as a step toward lung regeneration and disease modeling. Biomaterials 113: 118– 132
Crossref
CAS
PubMed
Web of Science®Google Scholar - 85. Tauriello DVF, Palomo‐Ponce S, Stork D, Berenguer‐Llergo A, Badia‐Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando‐Momblona X et al (2018) TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554: 538– 543
Crossref
CAS
PubMed
Web of Science®Google Scholar - 86. Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V et al (2005) Gefitinib plus best supportive care in previously treated patients with refractory advanced non‐small‐cell lung cancer: results from a randomised, placebo‐controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 366: 1527– 1537
Crossref
CAS
PubMed
Web of Science®Google Scholar - 87. Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, Froeling FEM, Burkhart RA, Denroche RE, Jang G et al (2018) Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov 8: 1112– 1129
Crossref
CAS
PubMed
Web of Science®Google Scholar - 88. Toolan HW (1953) Growth of human tumors in cortisone‐treated laboratory animals: the possibility of obtaining permanently transplantable human tumors. Cancer Res 13: 389– 394
CAS
PubMed
Web of Science®Google Scholar - 89. Verissimo CS, Overmeer M, Ponsioen B, Drost J, Mertens S, Verlaan‐klink I, Van Gerwen B, Van Der Ven M, Van De Wetering M, Egan DA et al (2016) Targeting mutant RAS in patient‐derived colorectal cancer organoids by combinatorial drug screening. Elife 5: e18489
Crossref
PubMed
Web of Science®Google Scholar - 90. Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández‐mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R et al (2018) Patient‐derived organoids model treatment response of metastatic gastrointestinal cancers. Science 926: 920– 926
Crossref
Web of Science®Google Scholar - 91. Voloshin T, Gingis‐Velitski S, Bril R, Benayoun L, Munster M, Milsom C, Man S, Kerbel RS, Shaked Y (2011) G‐CSF supplementation with chemotherapy can promote revascularization and subsequent tumor regrowth: prevention by a CXCR92 antagonist. Blood 118: 3426– 3435
Crossref
CAS
PubMed
Web of Science®Google Scholar - 92. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN et al (2013) Increased survival in pancreatic cancer with nab‐paclitaxel plus gemcitabine. N Engl J Med 369: 1691– 1703
Crossref
CAS
PubMed
Web of Science®Google Scholar - 93. Wang Y, Revelo MP, Sudilovsky D, Cao M, Chen WG, Goetz L, Xue H, Sadar M, Shappell SB, Cunha GR et al (2005) Development and characterization of efficient xenograft models for benign and malignant human prostate tissue. Prostate 64: 149– 159
Wiley Online Library
CAS
PubMed
Web of Science®Google Scholar - 94. Weeber F, van de Wetering M, Hoogstraat M, Dijkstra KK, Krijgsman O, Kuilman T, Gadellaa‐van Hooijdonk CGM, van der Velden DL, Peeper DS, Cuppen EPJG et al (2015) Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc Natl Acad Sci USA 112: 13308– 13311
Crossref
CAS
PubMed
Web of Science®Google Scholar - 95. Wege AK, Ernst W, Eckl J, Frankenberger B, Vollmann‐Zwerenz A, Männel DN, Ortmann O, Kroemer A, Brockhoff G (2011) Humanized tumor mice‐A new model to study and manipulate the immune response in advanced cancer therapy. Int J Cancer 129: 2194– 2206
Wiley Online Library
CAS
PubMed
Web of Science®Google Scholar - 96. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, Van Houdt W, Van Gorp J, Taylor‐Weiner A, Kester L et al (2015) Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161: 933– 945
Crossref
CAS
PubMed
Web of Science®Google Scholar - 97. Wimmer RA, Leopoldi A, Aichinger M, Wick N, Hantusch B, Novatchkova M, Taubenschmid J, Hämmerle M, Esk C, Bagley JA et al (2019) Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565: 505– 510
Crossref
CAS
Web of Science®Google Scholar - 98. Yan HHN, Siu HC, Law S, Ho SL, Yue SSK, Tsui WY, Chan D, Chan AS, Ma S, Lam KO et al (2018) A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 23: 882– 897
Crossref
CAS
Web of Science®Google Scholar - 99. Yuan Z, Chen S, Sun Q, Wang N, Li D, Miao S, Gao C, Chen Y, Tan C, Jiang Y (2017) Olaparib hydroxamic acid derivatives as dual PARP and HDAC inhibitors for cancer therapy. Bioorganic Med Chem 25: 4100– 4109
Crossref
CAS
PubMed
Web of Science®Google Scholar - 100. Zhan B, Wen S, Lu J, Shen G, Lin X, Feng J, Huang H, Zhan B, Wen S, Lu J et al (2017) Identification and causes of metabonomic difference between orthotopic and subcutaneous xenograft of pancreatic cancer. Oncotarget 8: 61264– 61281
CrossrefGoogle Scholar - 101. Zhang C, Yang C, Feldman MJ, Wang H, Pang Y, Maggio DM, Zhu D, Nesvick CL, Dmitriev P, Bullova P et al (2017) Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 8: 56110– 56125Google Scholar
- 102. Zumwalde NA, Haag JD, Sharma D, Mirrielees JA, Wilke LG, Gould MN, Gumperz JE (2016) Analysis of immune cells from human mammary ductal epithelial organoids reveals Vδ2+T cells that efficiently target breast carcinoma cells in the presence of bisphosphonate. Cancer Prev Res 9: 305– 316
Crossref
CAS
Web of Science®Google Scholar